Endoplasmic Reticulum Stress and Metabolism
Metabolic Disease
We aim to understand the relationship between stress in the secretory pathway, the unfolded protein response and fatty liver.
Research Questions
- What are the mediators of UPR induced fatty liver?
- How does Atf6 activation cause steatosis?
- What differentiates between a stressed and adaptive UPR?
- Can an adaptive UPR be harnessed to protect against fatty liver?
Fatty liver disease (FLD) is emerging as a global health crisis, as obesity, a main FLD etiology, afflicts over 13% of the world population. Additionally, alcohol, toxins and inherited metabolic diseases cause FLD, making this the most prevalent liver disease globally. Shared cellular responses to hepatic lipid overload include inflammation, cell damage and death, the magnitude of which may dictate the clinical outcome. Recent, exciting data shows that lipid accumulation in hepatoctyes (steatosis) from diverse etiologies is accompanied by activation of the unfolded protein response (UPR), a quality control mechanism that assures rapid and efficient processing of proteins through the secretory pathway. Mounting evidence implicates the UPR as a key mediator in the development and progression of FLD. The mechanism by which UPR activation causes FLD is a major, unanswered question in the field and a central focus of research in the Sadler Laboratory.
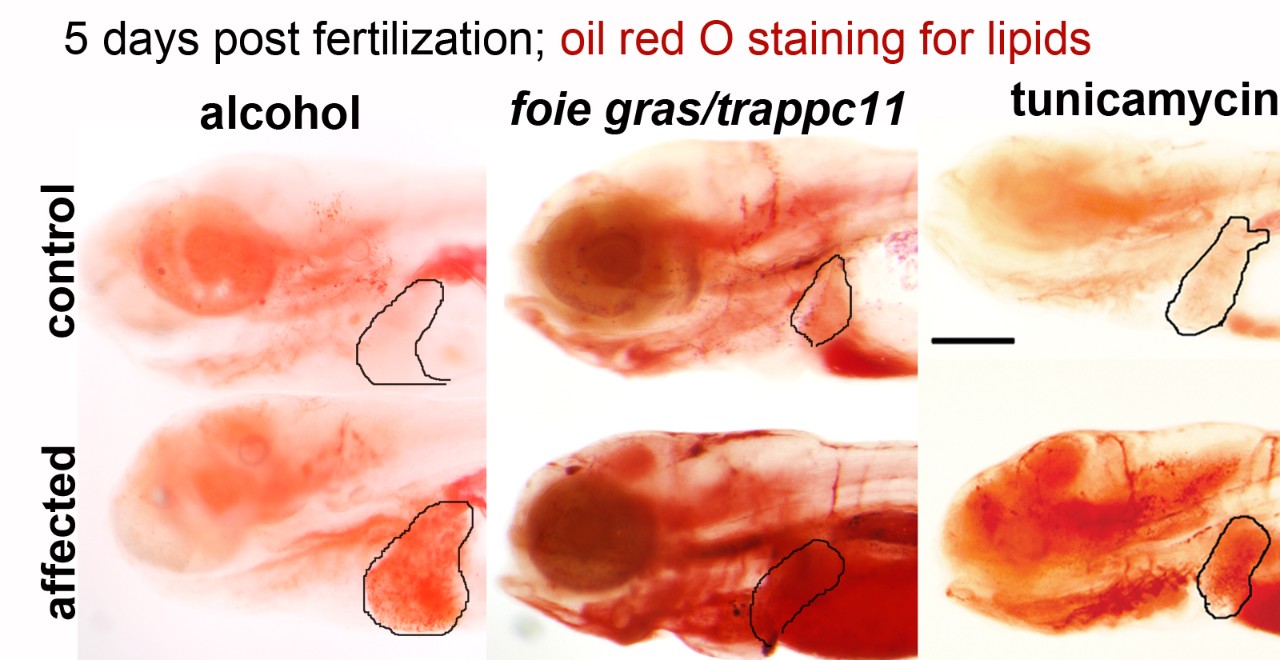

Using genetics, genomics and imaging approaches in zebrafish, we discovered some stress induces a UPR that cause FLD, other UPRs protect against it. The novelty of our work is in the system we use (zebrafish) and our integrated experimental approaches (genetics, genomics, cell biology) and the importance of the disease we study. Our approach is interdisciplinary, as we have merged our expertise in foundation in developmental and cell biology with a clinical problem that is a pressing public health concern.
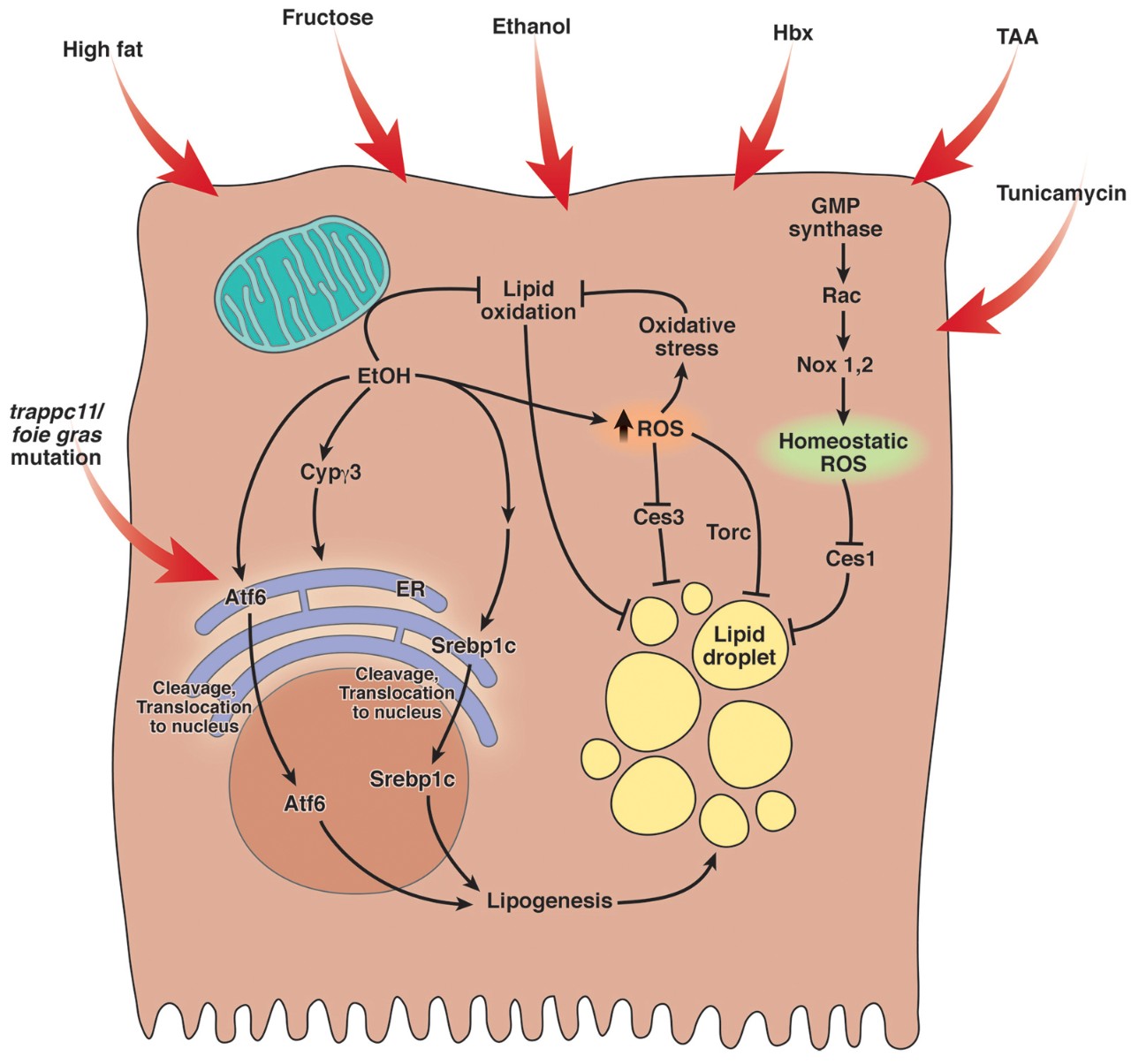
Our aim is to identify signatures of “stressed” and “adaptive” UPR subclasses with the goal of silencing the pathological aspects of the UPR and inducing the protective aspects as a means to treat FLD. Our work has identified the Atf6 transcription factor as necessary and sufficient for causing FLD and thus is a central component of the stressed, disease causing UPR.
There are implications to our work beyond FLD. The UPR is required for the homeostasis of cells that maintain a high volume secretory pathway. Additionally, the UPR is activated in non-secretory cells in response to pathological insults, such as oxidative damage, that compromise endoplasmic reticulum (ER) or Golgi function. Several diseases, including cystic fibrosis, á-1 antitrypsin deficiency, Parkinson’s, and Alzheimer’s are attributed to protein misfolding.
In these cases, UPR activity is not sufficient to manage the unfolded protein load, resulting in protein aggregates, failure to deliver the mutant protein to its correct location in the cell, and ultimately, cell death. Recent therapeutic strategies for cystic fibrosis and á-1 antitrypsin deficiency target the UPR, and an anticipated outcome of our work is to identify drugs that augment this process. If successful, these compounds could also yield progress in treating other diseases of protein misfolding.