Research
Molecular Machines
Role of Conformational Transitions on Enzyme Specificity
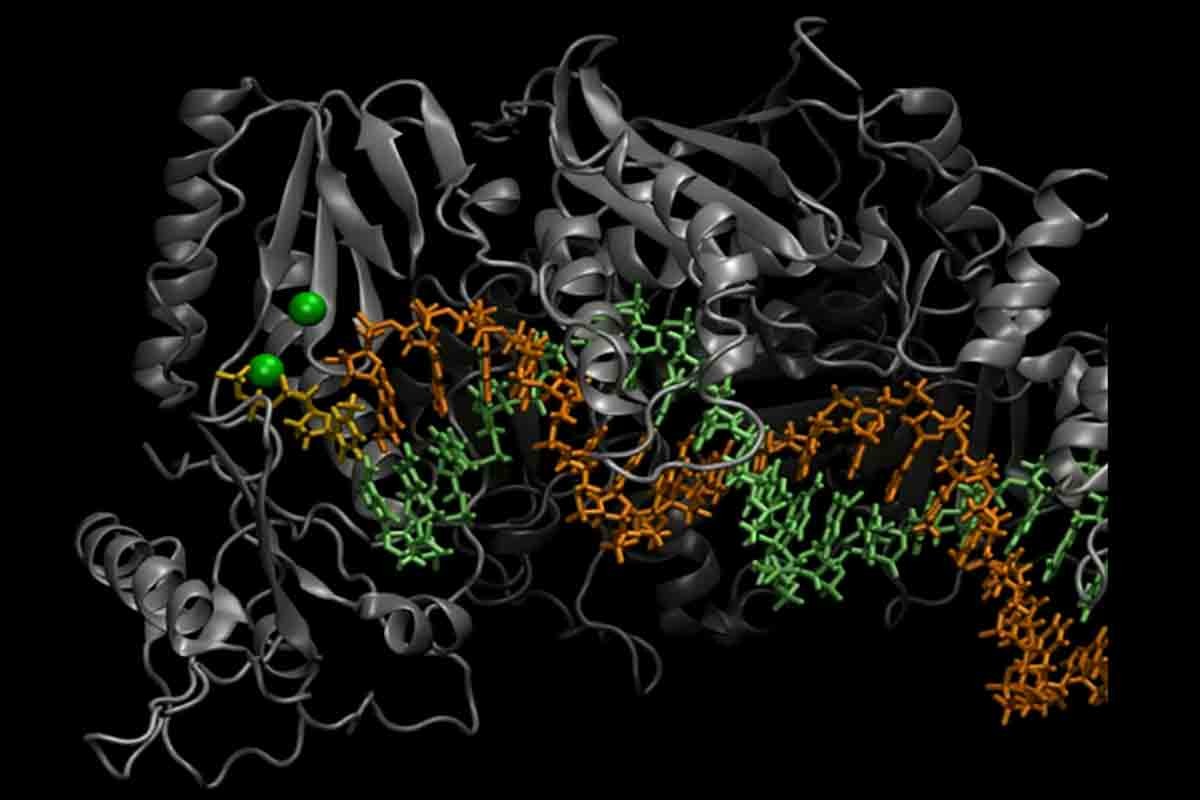

Molecular machines (MM) are essential components of living cells. They conduct mechanical work, carry cargo, assist in enzymatic reactions, and more. Our lab is interested in deciphering the physical and chemical mechanisms by which these fascinating machines function. MM operate by making conformational transitions. For example closing of the fingers domain of the HIV Reverse Transcriptase, an enzyme that is responsible for replicating the DNA of HIV virus shown in the movie above is one of these conformational transitions. Connection between the thermodynamics and kinetics of these transitions to their operation is of great importance in biology and bioengineering. Particularly, we are interested in the role of conformational transitions on the fidelity of molecular machines. Our current focus is on three biomolecular systems: DNA polymerase, Ribozymes and Hexokinase.
RNA Folding
Atomically detailed simulation of kinetics and Thermodynamics
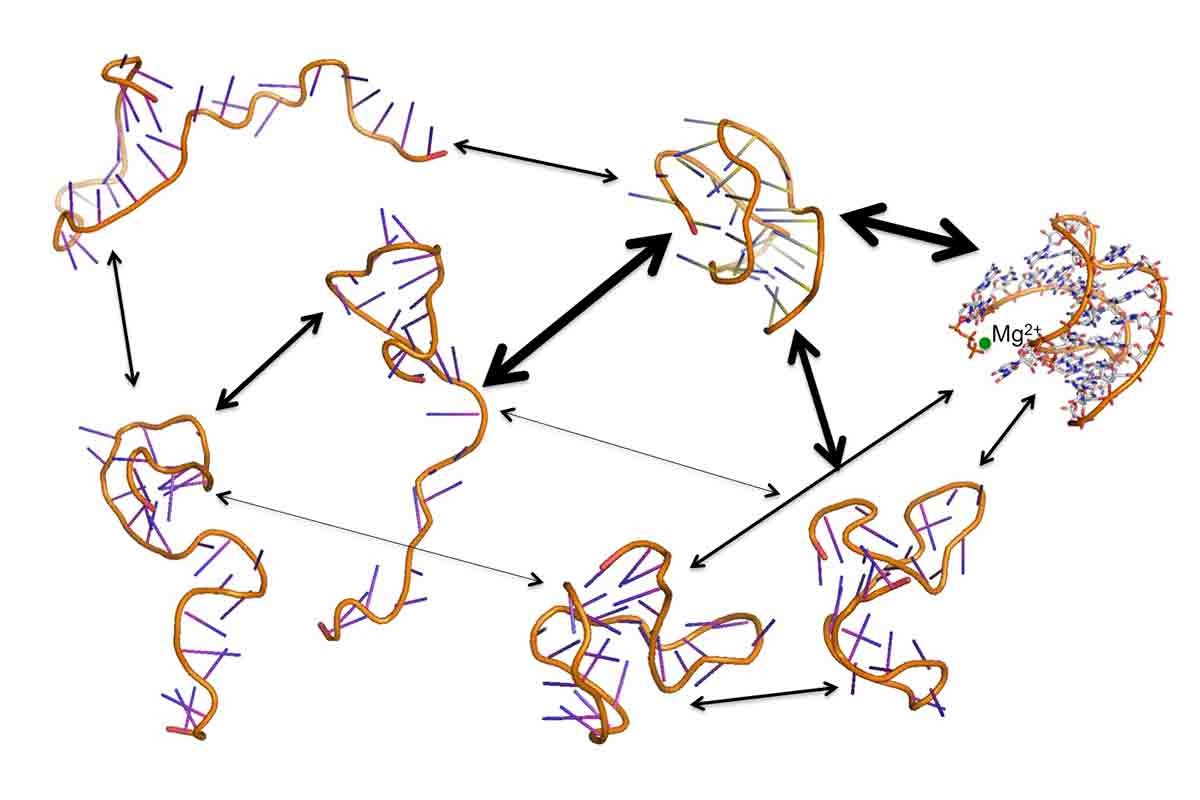
RNA folding is one of the most intriguing problems of computational biology and hence many groups are interested in studying it both experimentally and theoretically. Shown in the Figure above is a schematic folding pathway of Beet Western Yellows Virus (BWYV) generated by unfolding the RNA with high temperature Molecular Dynamics simulations. There are many fundamental questions on RNA folding, for example how does the negatively charged polymer chain folds into a well defined three-dimensional structure? What is the role of counter ions on the folding kinetics and thermodynamics? How do the pathways affect by different ionic conditions? Atomically detailed simulations are needed to understand these fundamental questions. However such simulations are very challenging since folding kinetics of RNA folding is in seconds, far beyond accessible time scale with straightforward Molecular Dynamics simulations (microseconds). In addition folding process involves multiple pathways and many intermediate states. A true kinetics study would require hundreds of folding events to simulate the reality while even one event is hardly accessible with current technology.
We develop and use multi-scale computational modeling approaches to study this process. The idea relies on exploring different regions of configuration space independently by computing short trajectories and glue them using a theory called Milestoning.